Genomic Pipeline on 1,000 CPUs
Genomic Pipeline on 1,000 CPUs
In this example we:
- Download raw Illumina sequencing data from GSE165845.
- Download the reference genome plus the Illumina BPM and EGT files.
- Convert 360 samples in parallel, then merge everything into PGEN/PVAR/PSAM files.
This is a pretty normal genomics workflow. The weird part is usually not the science. The weird part is getting all the command-line tools, reference files, sample files, and CPUs lined up at the same time.
Step 1: Boot some VMs
For this run we boot 13 VMs with 80 CPUs each.
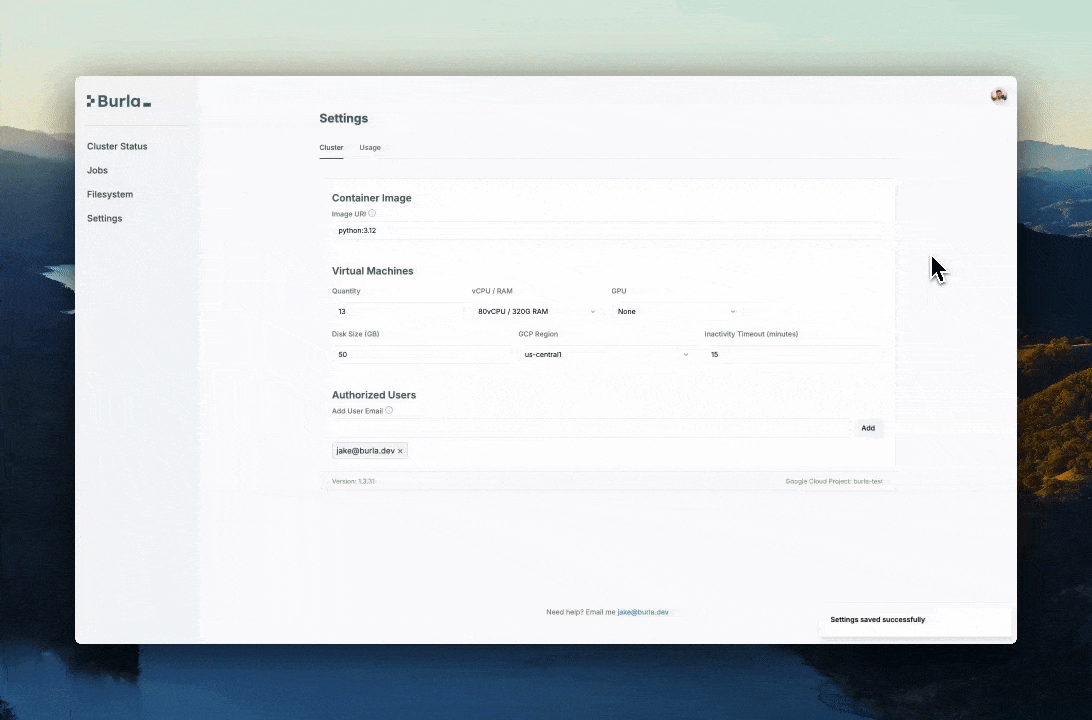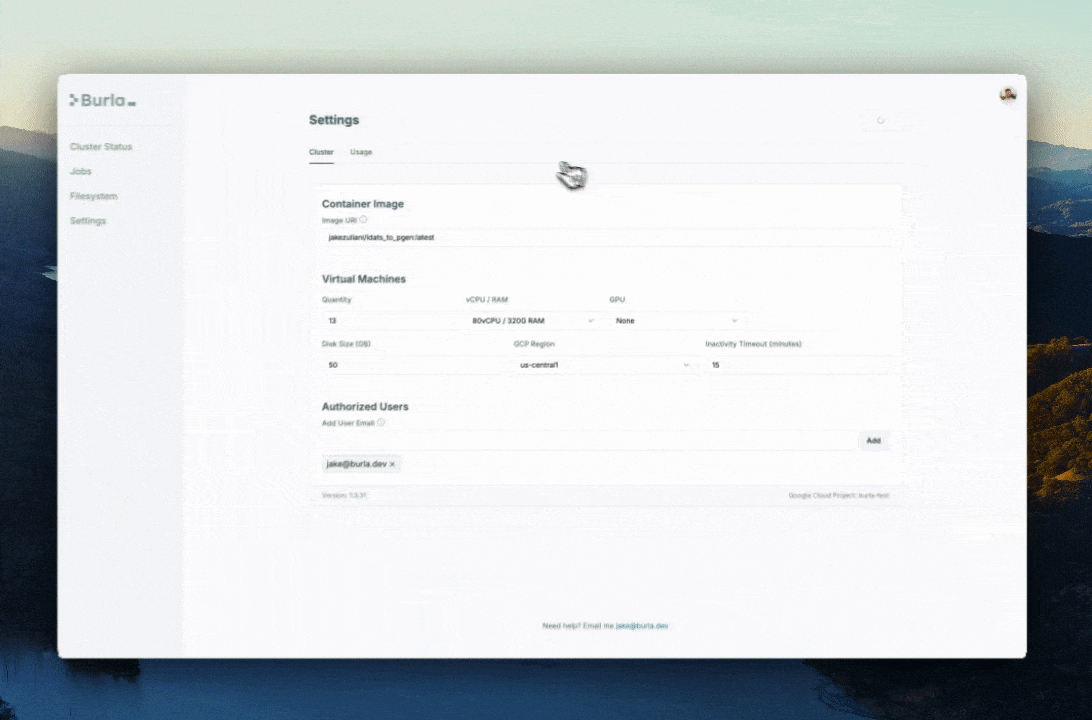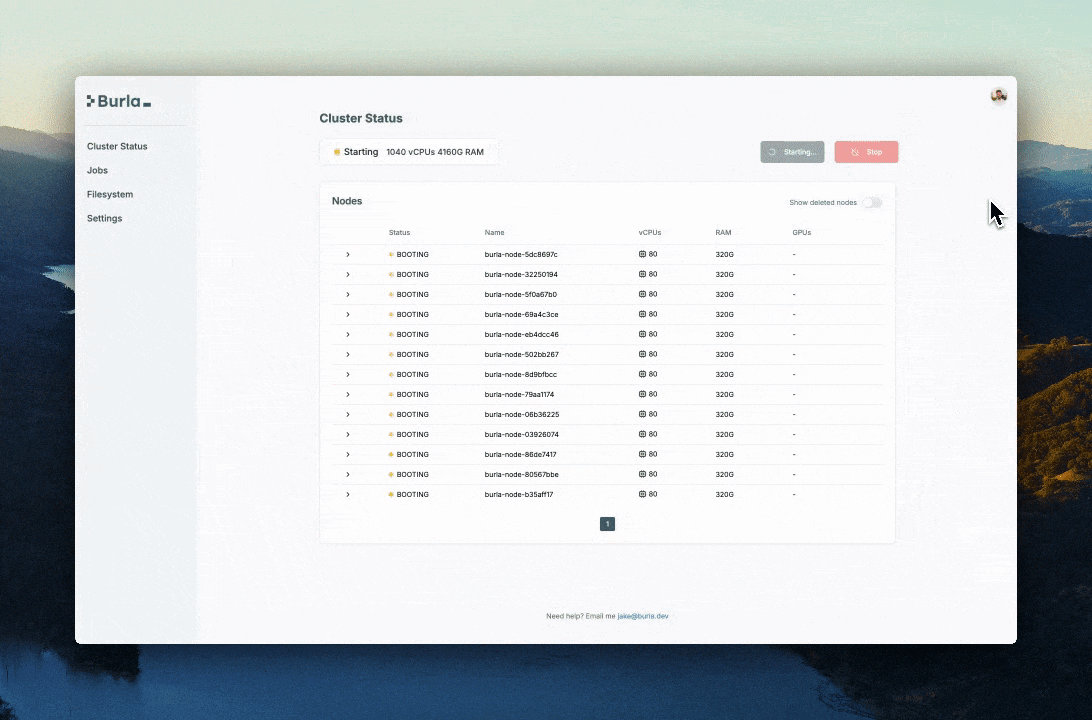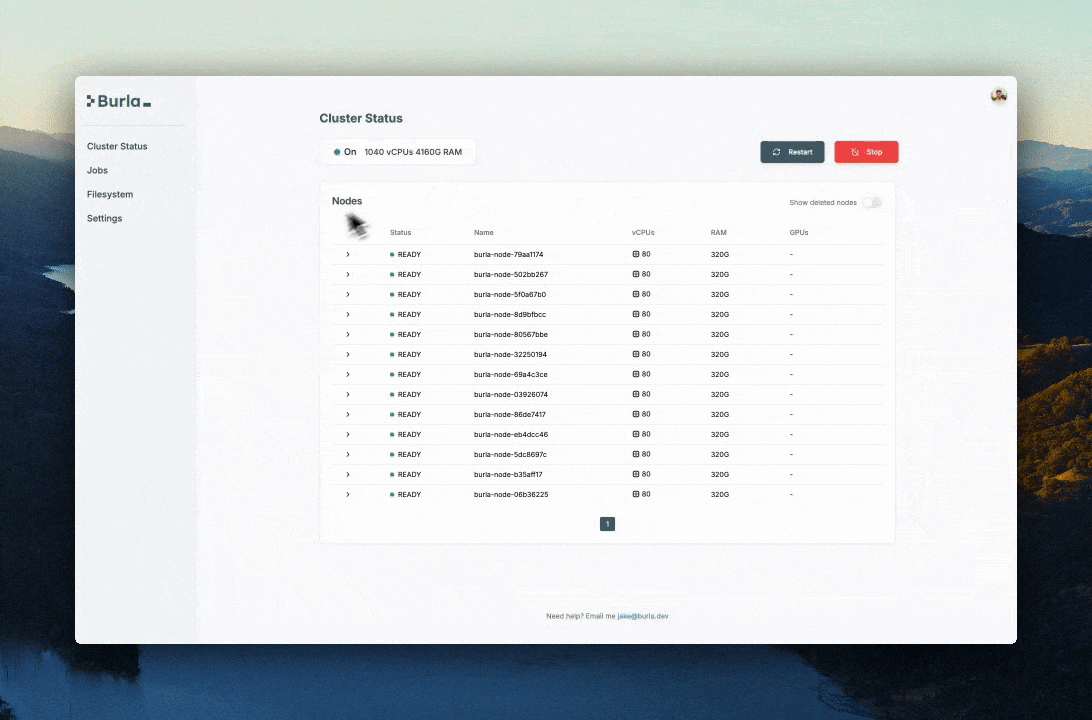
We use a custom Docker image:
jakezuliani/idats_to_pgen:latestThat image has bcftools, plink, and plink2 installed. Burla runs our Python functions inside that image, so the worker can call the same tools I would call from a terminal.
Step 2: Download prerequisite data
First we download the reference genome, the BPM file, and the EGT cluster file. Everything goes into ./shared, which is backed by a Google Cloud Storage bucket.
Imports and URL definitions
import io
import gzip
import zipfile
import requests
import subprocess
from shutil import move
from pathlib import Path
import pandas as pd
from burla import remote_parallel_map
def _run_cmd(cmd: str, print_output=False):
kwargs = {} if print_output else dict(stdout=subprocess.PIPE, stderr=subprocess.PIPE)
process = subprocess.run(cmd, shell=True, text=True, **kwargs)
if process.returncode != 0:
print(process.stdout)
print(process.stderr)
raise subprocess.CalledProcessError(process.returncode, cmd)
SAMPLE_INFO_URL = "https://storage.googleapis.com/burla-demo-data/genomic_sample_info.csv"
REF_GZ_URL = "ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/human_g1k_v37.fasta.gz"
REF_FAI_URL = "ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/human_g1k_v37.fasta.fai"
illumina_base_url = "https://webdata.illumina.com/downloads/productfiles/global-screening-array/v1-0"
BPM_URL = f"{illumina_base_url}/infinium-global-screening-array-v1-0-c1-manifest-file-bpm-build37.zip"
CLUSTER_URL = f"{illumina_base_url}/infinium-global-screening-array-v1-0-c1-cluster-file.zip"
REF_PATH = Path("shared/idat_to_pgen_pipeline/human_g1k_v37.fasta")
REF_GZ_PATH = REF_PATH.with_suffix(".fasta.gz")
REF_FAI_PATH = REF_PATH.with_suffix(".fasta.fai")
BPM_PATH = Path("shared/idat_to_pgen_pipeline/GSA-24v1-0_C1.bpm")
EGT_PATH = Path("shared/idat_to_pgen_pipeline/GSA-24v1-0_C1_ClusterFile.egt")def download_prerequisite_data(_):
Path("shared/idat_to_pgen_pipeline").mkdir(parents=True, exist_ok=True)
if not BPM_PATH.exists():
bpm_zip = zipfile.ZipFile(io.BytesIO(requests.get(BPM_URL).content))
cluster_zip = zipfile.ZipFile(io.BytesIO(requests.get(CLUSTER_URL).content))
bpm_zip.extractall("shared/idat_to_pgen_pipeline")
cluster_zip.extractall("shared/idat_to_pgen_pipeline")
if not REF_PATH.with_suffix(".fasta.gz").exists():
_run_cmd(f"wget --passive-ftp -O {REF_GZ_PATH} {REF_GZ_URL}", print_output=True)
_run_cmd(f"truncate -s 891946027 {REF_GZ_PATH}", print_output=True)
_run_cmd(f"gunzip -f {REF_GZ_PATH}", print_output=True)
if not REF_FAI_PATH.exists():
_run_cmd(f"samtools faidx {REF_PATH}", print_output=True)
df = pd.read_csv(SAMPLE_INFO_URL)
return list(zip(df.sample_id, df.cell_line, df.replicate))
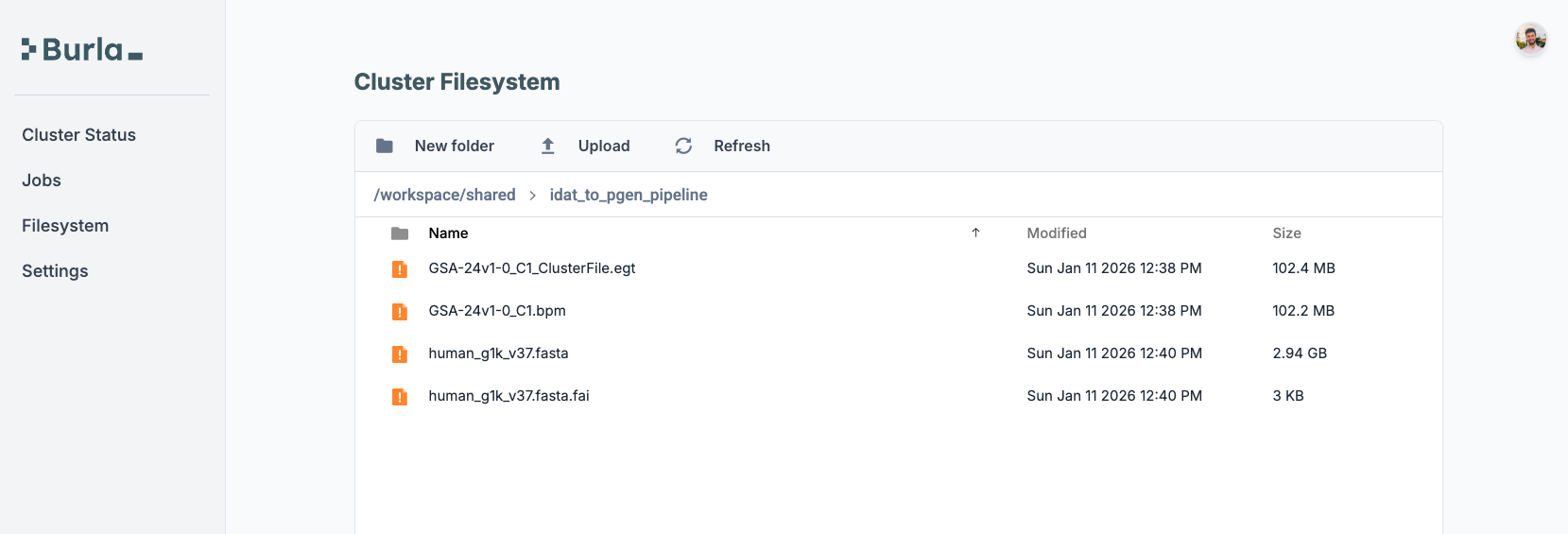
sample_info = remote_parallel_map(download_prerequisite_data, [None])[0]After that, the prerequisite files show up in the dashboard filesystem.
Step 3: Download the IDAT files
Each sample has a red and green IDAT file, so 360 samples means 720 files. This is exactly the kind of thing I don't want to do one at a time.
def download_single_idat_file(_id, cell_line, replicate, color):
idat_path = Path(f"shared/idat_to_pgen_pipeline/{_id}/{cell_line}_{replicate}_{color}.idat")
idat_path.parent.mkdir(parents=True, exist_ok=True)
if not idat_path.exists():
url = f"https://www.ncbi.nlm.nih.gov/geo/download/?acc={_id}"
url += f"&format=file&file={_id}%5F{cell_line}%5F{replicate}%5F{color}%2Eidat%2Egz"
with gzip.open(io.BytesIO(requests.get(url).content)) as f_in, idat_path.open("wb") as f_out:
f_out.write(f_in.read())
sample_info_colored = [(*a, color) for a in sample_info for color in ("Grn", "Red")]
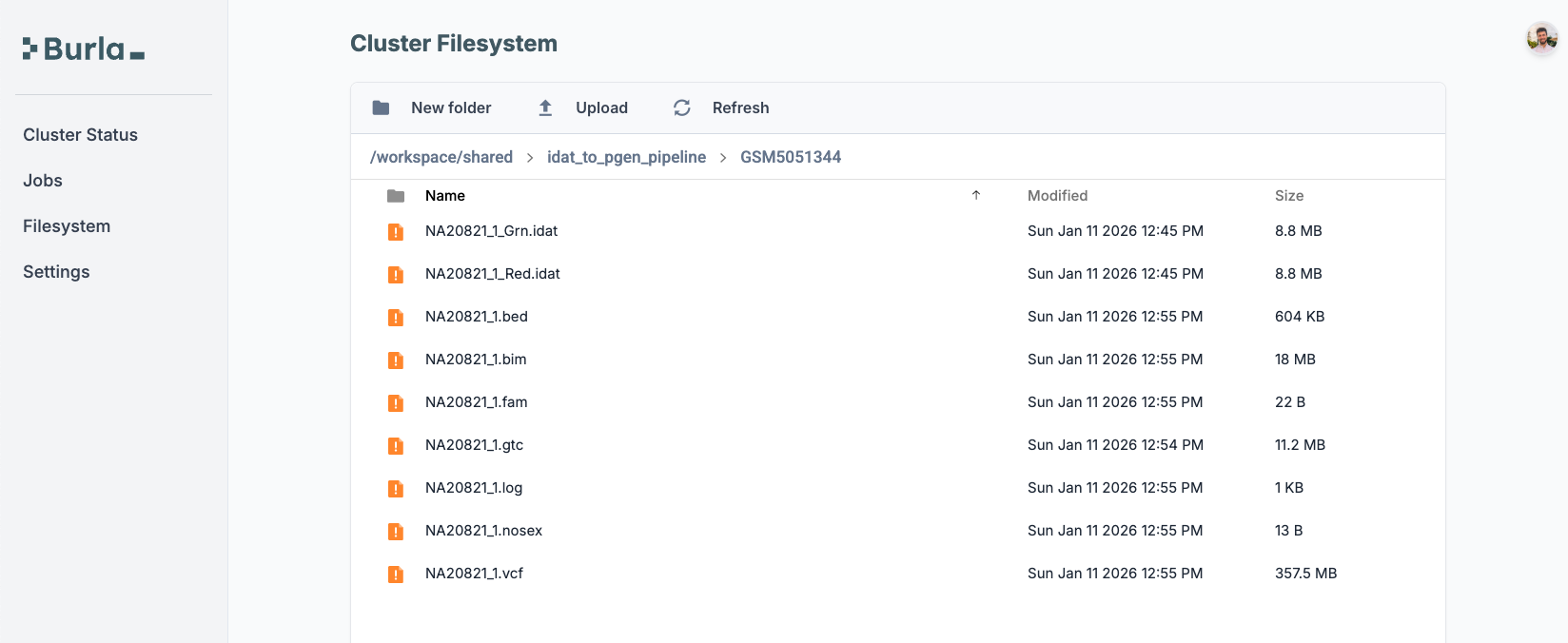
remote_parallel_map(download_single_idat_file, sample_info_colored)The sample folders now contain the red and green IDATs.
.gif)
Step 4: Convert every sample
Now each worker takes one sample, converts the IDAT pair to GTC, aligns to the reference genome, filters biallelic variants, and writes PLINK BED/BIM/FAM files.
def convert_idats_to_plink_bed_format(_id, cell_line, replicate):
gtc_path = Path(f"shared/idat_to_pgen_pipeline/{_id}/{cell_line}_{replicate}.gtc")
vcf_path = gtc_path.with_suffix(".vcf")
_run_cmd(f"bcftools +idat2gtc --bpm {BPM_PATH} --egt {EGT_PATH} --idats shared/idat_to_pgen_pipeline/{_id}", print_output=True)
move(f"{cell_line}_{replicate}.gtc", gtc_path)
_run_cmd(f"bcftools +gtc2vcf {gtc_path} -b {BPM_PATH} -e {EGT_PATH} -f {REF_PATH} -o {vcf_path} --do-not-check-bpm", print_output=True)
filtered_vcf_path = vcf_path.with_suffix(".temp_filtered.vcf")
_run_cmd(f"bcftools view -m2 -M2 -o {filtered_vcf_path} {vcf_path}", print_output=True)
filtered_vcf_path.rename(vcf_path)
_run_cmd(f"plink --vcf {vcf_path} --out {vcf_path.with_suffix('')} --const-fid 0, --make-bed --allow-extra-chr --keep-allele-order", print_output=True)
remote_parallel_map(convert_idats_to_plink_bed_format, sample_info, func_cpu=8)Each function call gets 8 CPUs and 32GB of RAM. That is enough for one sample, and it lets the whole cohort move at once.
Step 5: Merge the cohort
The final merge runs once, with a bigger worker.
MERGED_PATH = "shared/idat_to_pgen_pipeline/merged"
def combine_bed_files_into_pgen_file(sample_info):
mergelist = [f"shared/idat_to_pgen_pipeline/{_id}/{cl}_{r}" for _id, cl, r in sample_info]
mergelist_path = Path("merge_list.txt")
mergelist_path.write_text("\n".join(mergelist))
cmd = f"plink --bfile {mergelist[0]} --merge-list {mergelist_path} --out {MERGED_PATH} --allow-extra-chr --biallelic-only strict"
_run_cmd(cmd, print_output=True)
_run_cmd(f"plink2 --bfile {MERGED_PATH} --out {MERGED_PATH} --make-pgen --allow-extra-chr", print_output=True)
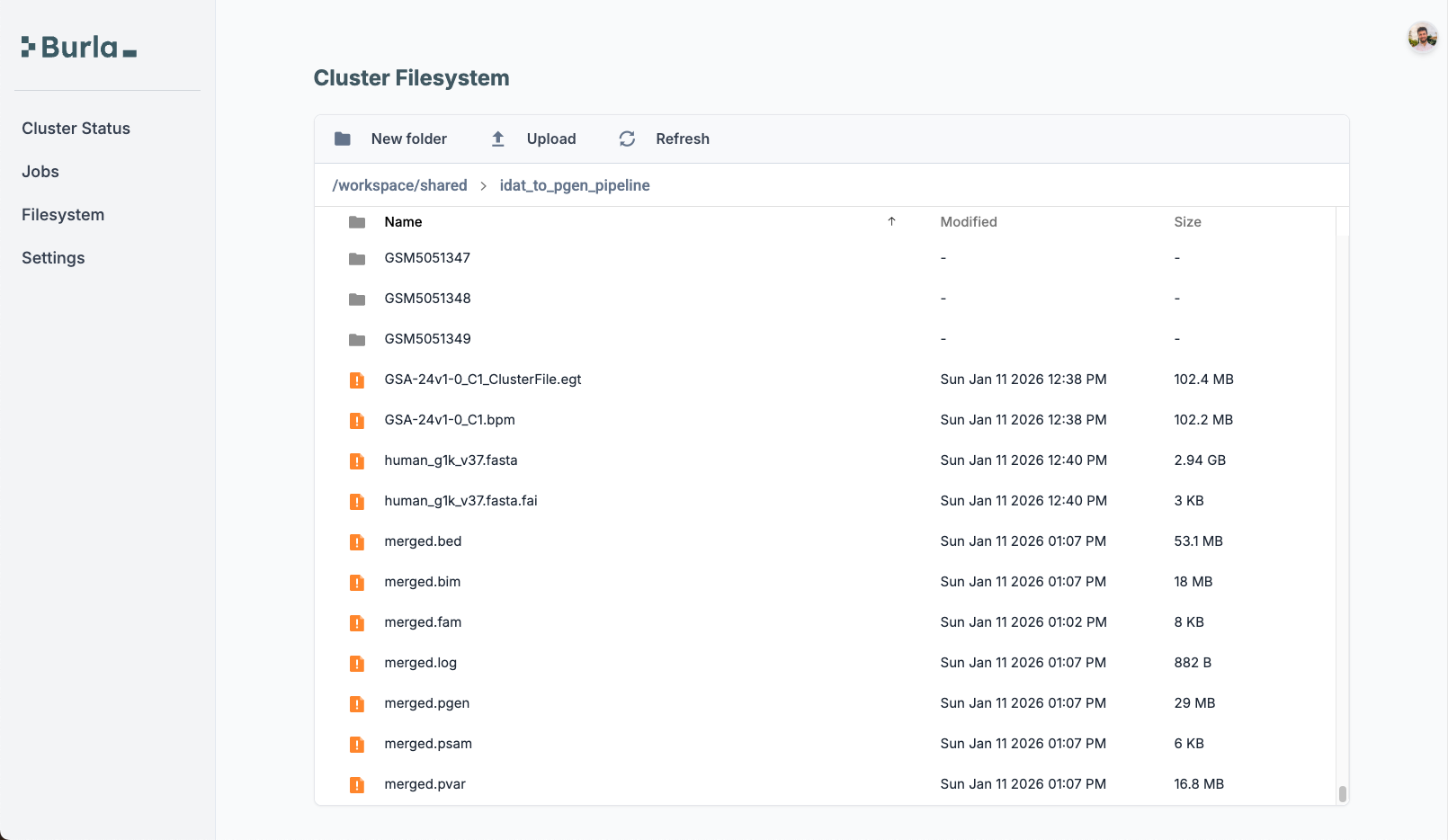
_ = remote_parallel_map(combine_bed_files_into_pgen_file, [sample_info], func_cpu=80, func_ram=320)After it finishes, the PGEN/PVAR/PSAM files are available in the dashboard.
Want to run this code yourself?
The demo is available as a Colab notebook:
https://colab.research.google.com/drive/1lEbeGOoowZ9FKA9yctziWyhH6TvLuxTi?usp=sharing
What's the point?
This is the real reason I like this example: the pipeline stays boring.
The worker code is still Python calling bcftools and plink. The files still live in a normal shared folder. The only real change is that instead of nursing one sample through the pipeline, we run the cohort at the size it was meant to run.
If this were my analysis, I would not want to spend the day building a scheduler before I even know whether the genotype conversion works. I would want to run the thing, look at the outputs, and only optimize once there is a real bottleneck.